Approximately 38 million people die from noncommunicable diseases (NCDs) globally per year (Global status report on noncommunicable diseases 2014: Attaining the nine global noncommunicable diseases targets; a shared responsibility, 2014), and the most important contributing factor to these deaths is cardiovascular disease. Cardiovascular diseases have become the top 3 causes of death not only in Taiwan but also worldwide ("World Health Organization website [http://www.who.int/mediacentre/factsheets/fs310/en/], accessed date: 2018/02/09,"). Notably, approximately 17.7 million people die from cardiovascular diseases annually, which causes inestimable socioeconomic losses. Coronary artery disease is one of the most common cardiovascular diseases associated with poorer prognosis in patients. It is well known that plaques resulting from atherosclerosis cause coronary arteries to narrow, which may eventually lead to a heart attack and/or sudden death. Therefore, many people are focused on sound eating behaviors, a healthy lifestyle and precise weight management to avoid coronary artery blockage. However, as the opposite of coronary artery blockage, a relatively rare disease called coronary artery ectasia (CAE) is characterized by abnormally dilated coronary arteries (Figure 1). The criterion used to define CAE is dilation of a coronary artery to 1.5 times the diameter of an adjacent normal vessel (Al-Tamimi & Al-Dhuhli, 2009; Dahhan, 2015). The incidence rate of CAE is approximately 5% worldwide (Huang, Liu, Chen, & Li, 2014), which suggests that CAE is a rare clinical condition. Although the incidence rate of CAE is low, CAE patients may suffer from angina, myocardial ischemia, myocardial infarction and, in the worst case, heart failure (Mavrogeni, 2010). The pathogenic mechanism of CAE is still unclear, and treatment for CAE is limited. Therefore, it is important to provide a better understanding of the mechanism of CAE to facilitate identification of its possible biomarkers and/or drug targets (Lu et al., 2017). Previous studies have demonstrated that smoking, inflammation, and hyperlipidemia are three major risk factors for CAE (Boles, Zhao, David, Eriksson, & Henein, 2012; Li, Li, & Li, 2007; Li, Nie, Qian, Zeng, & Zhang, 2009; Yang et al., 2013), and DNA methylation has been reported as a potential driver of these risk factors (Guay et al., 2012; Kumar et al., 2013; Lee & Pausova, 2013; Stenvinkel et al., 2007; Suzuki, Toyota, Kondo, & Shinomura, 2009). DNA methylation is a modification of the DNA structure of a gene by the addition of an additional methyl group, without changing the DNA sequence. The mRNA expression level of a gene is usually inversely related to its DNA methylation level, i.e., a gene is down-regulated if its DNA is hypermethylated. To date, no systematic DNA methylation studies related to CAE have been reported. Therefore, we aimed to investigate the genome-wide methylation profiles of CAE patients. A total of 12 CAE patients and 12 matched controls recruited from National Taiwan University Hospital were studied by using the Illumina HumanMethylation450k microarray. A propensity score was calculated to select appropriate controls with clinical and demographic characteristics similar to those of the CAE patients. After analyzing all the genes, the Wilcoxon rank sum test identified 89 genes that showed significantly different methylation levels between the CAE patients and controls (P value<0.05 and Δβ >|0.1|). Two approaches, including over-representation analysis and gene set enrichment analysis, were utilized to understand which biological functions and signaling pathways were affected based on the methylation levels. Notably, both approaches indicated that the genes involved in immune and inflammatory responses had dysregulated methylation levels. To validate the microarray results, we selected 6 genes (NOTCH4, HLA-DRB1, C3, HLA-DQA1, TLR6, and PENK) that showed the greatest difference between the case and control groups and validated them by using pyrosequencing experiments in 29 CAE patients and 89 matched controls. Among these 6 genes, TLR6 and NOTCH4 showed significant differences in methylation levels in both the microarray and pyrosequencing experiments. In general, the results of the pyrosequencing experiments revealed the same patterns as those demonstrated by the methylation microarrays, suggesting that the methylation changes are reproducible even when the detection method is changed. Finally, we found that the protein expression level of TLR6 was significantly lower in the CAE patients according to enzyme-linked immunosorbent assay (ELISA) data, which suggests that the DNA hypermethylation of TLR6 led to a lower expression of its downstream protein product. In conclusion, this genome-wide methylation study showed that several genes, including TLR6, had significantly different methylation levels in CAE patients. Thus, further investigations are warranted to elucidate their functional roles in the pathogenic process.
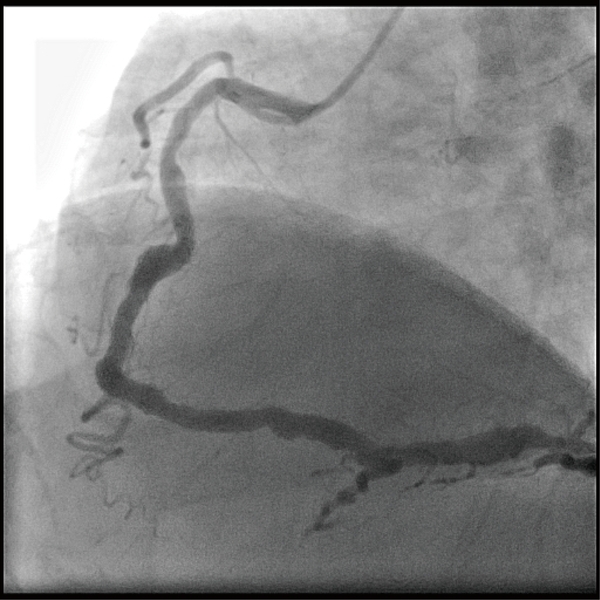
Figure 1. A representative photo of the dilated coronary artery in a CAE patient.
Reference
Lu, T., Chuang, N., Cheng, C., Hsu, C., Wang, Y., Lin, Y., . . . Juang, J. (2017). Genome-wide methylation profiles in coronary artery ectasia. Clinical Science, 131(7), 583-594. DOI:10.1042/CS20160821
Tzu-Pin Lu
Assistant Professor, Institute of Epidemiology and Preventive Medicine, National Taiwan University
Jyh-Ming Jimmy Juang
Clinical Associate Professor, Cardiovascular Center and Division of Cardiology, Department of Internal Medicine, National Taiwan University Hospital